
2024.04.25
未診断小児疾患の患者家族が辿った道:原因遺伝子の特定とiPS技術の応用可能性
遺伝子の一塩基が違うだけで、てんかん発作や重度運動障害で死に至る難病がある。
「NEDAMSS」と呼ばれる新規疾患に、一人の研究者が果敢に挑んでいる。
獨協医科大学の井上健一准教授は、ある家族の願いがきっかけでこの難病の謎解きに乗り出した。
孤発性IRF2BPL変異によるNEDAMSS病態モデルの確立に焦点を当て、未知の遺伝子変異から病態の解明、治療法開発の可能性までを追う。
未知の難病に光を当てる研究が向かう未来とは?
科学的探究の舞台裏とその社会的意義を紐解いていくインタビュー。

インタビュイー
井上 健一氏
獨協医科大学 先端医科学研究センター
准教授
■受賞歴
とちぎテックプラングランプリ Real Tech Fund賞
獨協医学会学術集会最優秀賞(第43回、49回)
とちぎテックプラングランプリ Real Tech Fund賞
獨協医学会学術集会最優秀賞(第43回、49回)
ある母親の願いから始まった難病研究

―――まずは、研究領域について教えてください。
NEDAMSSというのは、希少疾患の一種です。正確には英語で、「NEURODEVELOPMENTAL DISORDER WITH REGRESSION, ABNORMAL MOVEMENTS, LOSS OF SPEECH, AND SEIZURES」という名前がつけられています。この疾患は、ALSという病気に類似しており、徐々に体の動きが制限され、最終的には言葉も失われ死に至る神経変性疾患です。多くの場合、幼児期に発症する小児疾患として知られています。
近年、この症状の原因遺伝子が特定され、新規疾患として分類がなされましたが、それまでは確定診断をすることができず、対処療法しかない状況でした。症状としては、てんかんなどが見られ、徐々に歩行や言語能力が低下するという特徴があります。
5年から6年前に、IRF2BPLという遺伝子の異常が原因であることが明らかになりました。この発見は、以前からこの病気に携わってきた家族、臨床医、研究者たちにとって大きな進展でした。
私自身は、この病気の患者やその家族と関わる機会があり、遺伝子の関与が明らかになる前から、この病気について研究を進めてきました。その過程で、IRF2BPL遺伝子が特に重要な役割を果たしていることが明らかになり、引き続きその研究を進めています。
NEDAMSSの発症数は、2018年にアメリカで約十数例、フランスで同様の数の症例が報告されました。ただし、これは当時、遺伝子検査が頻繁に行われている施設においてのみの報告であり、潜在的にはもっと多くの症例が存在する可能性があります。
日本では大規模な研究は行われていませんが、現在のシステムの一つで、医師が難病の原因を探るために、患者のゲノム全体を調査する研究が進められています。 その結果、さまざまな遺伝子変異が見つかることがありますが、その意味までは明らかにならないことも少なくありません。
しかし、IRF2BPLの重要性が判明した後、過去に研究した遺伝子変異に再度注目するよう促すと、日本でもこの変異がいくつかの症例で見つかったようです。
日本では確認されているだけでも3症例が、NEDAMSSと特定されています。
検査を行えば、まだ見つかっていない潜在的な症例が存在するはずです。この新規疾患は一般の小児科医には未だ認知されておらず、大学病院などを除けば積極的に遺伝子検査を実施する体制にはありません。将来的に厚生労働省がIRF2BPLの検査を保険適応すれば、同様の症例がさらに見つかる可能性はあります。
―――読者の皆様には、NEDAMSSという疾患に関して馴染みのない方も多いと思うのですが、NEDAMSSの概要や先生がこの疾患に取り組むきっかけとなった背景について、教えてください。

約10年前、元上司との繋がりから、歩行障害のような症状が出始めた香港系アメリカ人の男の子に関して連絡がありました。
母親は一心に子供の治療を望んでおり、各方面に問い合わせ、治療法を模索していたようです。その中で、当時新しく登場した次世代シーケンサーを用いて、子供のゲノム解析を実施しました。その結果、RUNX3、IRF2BPL、GSK3Aという三つの遺伝子に変異が見つかりました。
しかし、遺伝子の変異において、重要なのは変異の意味なのです。
新型コロナウイルスの例からも分かるように、変異には意味のあるものとないものがあります。悪性に影響する変異もあれば、感染性に影響を与えない変異もあります。
当時、この遺伝子の変異の解釈まではまだ明確ではありませんでした。
その中で、私が大学院生時代に研究していたRUNX3遺伝子のノックアウトマウスでは、運動障害や歩行障害などの神経症状が認められていました。
主治医がこの点に着目し、私の元上司を通して、私も研究に参画する運びとなりました。
そこで私たちは、この症例がRUNX3関連の新規疾患である可能性を考え、iPS細胞を樹立して研究を行うことにしました。上司が京都大学のiPS細胞研究所の井上治久教授に相談し、男の子から採血して、世界で初めてNEDAMSSに関するiPS細胞を樹立することができました。
その後、私はこのiPS細胞を用いて、当時まだ原因不明の疾患の病態解明研究を進めていきました。RUNX3が関与する可能性が最も高いと考えられていましたが、確証は持てていませんでした。
元々見つかった三つの遺伝子変異のうち、一つに絞り込むのは難しい作業でした。
従来であれば、疾患の家系をたどり、大規模で費用のかかる遺伝学的アプローチで研究する必要がありました。しかし、家族内に発症者がいない孤発例の場合、それは難しくなります。このような場合、アメリカのシステムはこの問題にアプローチするためのネットワークを構築しています。その一例が、未診断の遺伝子を特定するための「Undiagnosed Diseases Network」です。

このネットワークでは、進化の過程で保存されてきた相同性の高い遺伝子に注目します。
IRF2BPLという遺伝子は、ショウジョウバエのPitsという遺伝子と高い相同性があることが分かっており、機能面でも共通する部分があると考えられました。
補足すると、一般的に、人の遺伝子をショウジョウバエに戻してその機能を調査するという実験はよく行われます。一種の試験管のようなもので、ショウジョウバエは遺伝学の研究において非常に重要な役割を果たしており、これまで人類に多くの重要な発見をもたらしています。
このPitsという遺伝子が神経系の発生に関与があることが判明し、このことから、3つの候補遺伝子の中でIRF2BPLが最も患者の症状を説明できる可能性が高いと判断されたのです。
進化の過程で保存された相同性の高い遺伝子に着目するアプローチが、IRF2BPLを有力候補として浮かび上がらせた大きな手掛かりになり、これがIRF2BPL研究の出発点となりました。
難病NEDAMSSに挑む、iPS細胞の挑戦

―――研究が進む中で、疾患にアプローチできている部分や、症状が軽くなるといった研究の進展はありましたか?
残念ながら、先程対処療法について触れましたが、現時点で既存の治療法を変えるような進展はまだありません。
研究によって、アメリカを中心に民間の寄付金が集まり、複数の研究グループが薬の候補を見つける段階まで進んでいます。しかし、これらの薬を実際の患者に試験的に使用する段階にはまだ至っていません。
やはり既存の治療法に進展をもたらすためには、小児科医との連携が不可欠だと思います。
私の大きな目標は、小児科医の先生方が興味を持ち、臨床研究に参加する意義を理解して頂くことです。そのために、私自身も研究を通して、力を尽くさなければならないと考えています。
―――iPS細胞を用いた研究の利点は何でしょうか?他の研究方法と比較して、どのような優位性がありますか?
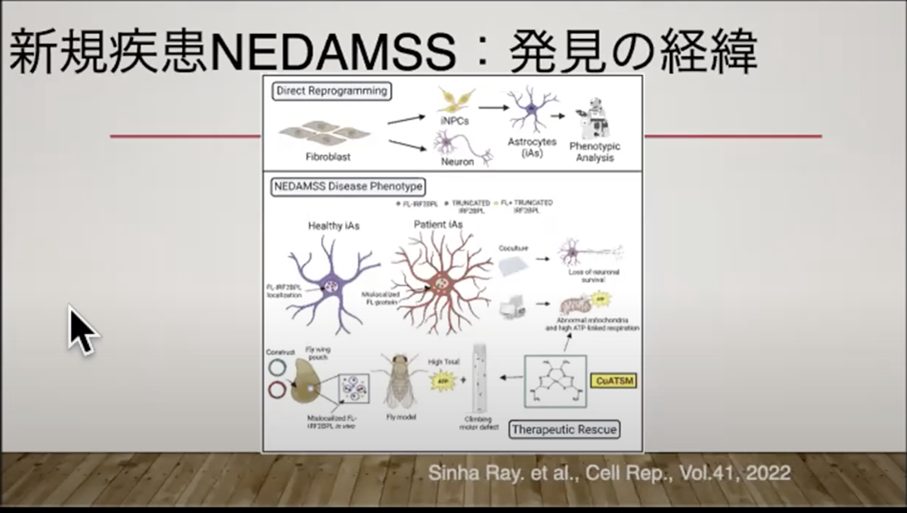
iPS細胞の作製には多くの準備とコストがかかりますが、その利点は科学的に厳密な結果が得られることにあります。
例えばアメリカでは、NEDAMSS患者さんの家族による寄付金で研究が進められていました。
その研究では、患者さんの皮膚細胞からアストログリアという細胞に直接分化転換させる手法が用いられました。アストログリアは脳内で神経細胞を支える重要な役割を担っています。この方法はiPS細胞技術と似ていますが、iPS細胞ほど細胞の品質管理が厳密ではありません。
具体的には、分化転換の過程で染色体が偶発的に欠損することがあり、そのようなデータの信頼性が低下してしまうリスクがあります。場合によっては、本来見られるべきでないデータ(False Positive)を発見してしまうこともあります。
一方、iPS細胞作製では時間はかかりますが、染色体の確認など細胞の品質管理が徹底されています。一人の人間に確実になれるようなiPS細胞だけを厳選して使用するため、より正確でデータの品質が高いのが特徴で、この点がiPS細胞の大きな強みです。
また、iPS細胞は一度作成すれば、ニューロンやアストログリア、さらには血液細胞などへの分化が可能です。
そのため、汎用性が高く、多くの組織に変化させることができる点でも、iPS細胞の優位性があります。
―――研究を進める上で、最も困難な問題は何でしたか?そして、それをどう乗り越えましたか?
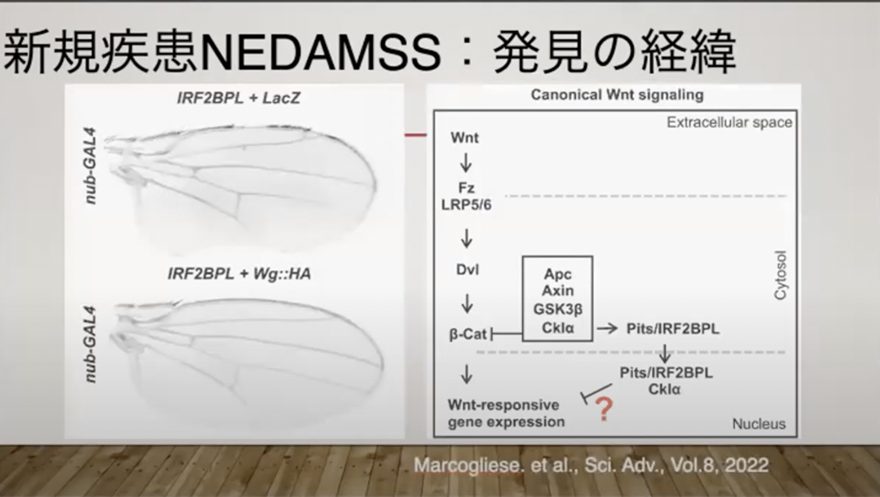
最初に、京都大学の井上教授と共同でNEDAMSSのiPS細胞を樹立したときは、うまくいかない時期がありました。
井上教授はALS研究の権威だったため、この患者さんの症状がALSと類似していることから、ALSの細胞表現型が見られるのではないかと考え、実験を行いました。
しかし、結果としてALSと類似した表現型は見られず、複数のプロジェクトを手がける多忙な教授と、見通しのない研究を続けることは困難を極めました。
それでもこの間に、別の日本人の患者さんのご家族と知り合い、もう一症例のiPS細胞を樹立したりしました。
そのような研究を進めているうちに、アメリカでアストログリアに異常があるらしいという論文が発表されました。
そこで気づいたのが、京都大学で使われていた方法は純粋なニューロンのみを分化誘導する技術は、実際の脳内環境とは異なるのではないかということです。アストログリアなどの周辺細胞が存在しない人工的な条件では、NEDAMSSの症状が再現されない可能性があると考えました。
そこで、私たちはニューロンとアストログリアを一緒に培養する新しい条件を模索し始めました。
現在、アストログリアに影響があることが分かってきたので、次はニューロンとアストログリアの共存在下でどのような影響を受けているかを解明することが課題となっています。
現在は、より脳内の自然な環境を再現した条件でiPS細胞を培養することが、今後の目標です。
iPS細胞技術は発見から18年が経過し*、日々進化を遂げています。特に神経系の研究分野では、iPS細胞がもたらす恩恵が大きいと期待されていました。
なぜなら、血液は患者から採取しやすく研究に用いられますが、脳組織を採取することは非常に難しいからです。頭蓋骨に守られた脳は、サンプルを得るために大がかりな手術が必要となり、研究が極めて困難でした。
しかし、iPS細胞技術の出現により、受精卵から種々の細胞や組織を作り出すことが可能になりました。つまり、実験室内で脳組織を再現することができるようになったのです。この革新的な技術は、神経系研究者にとって大きな福音となりました。
現在、iPS細胞の技術は日進月歩で進化しています。私たちも最新の技術動向をキャッチアップし、有用なものは積極的に取り入れていきたいと考えています。
参照:京都大学iPS細胞研究所「iPS細胞とは?」遺伝子から紐解く新たな可能性

―――将来的に、NEDAMSSの患者さんにどのような治療を提供できるようになると思いますか?
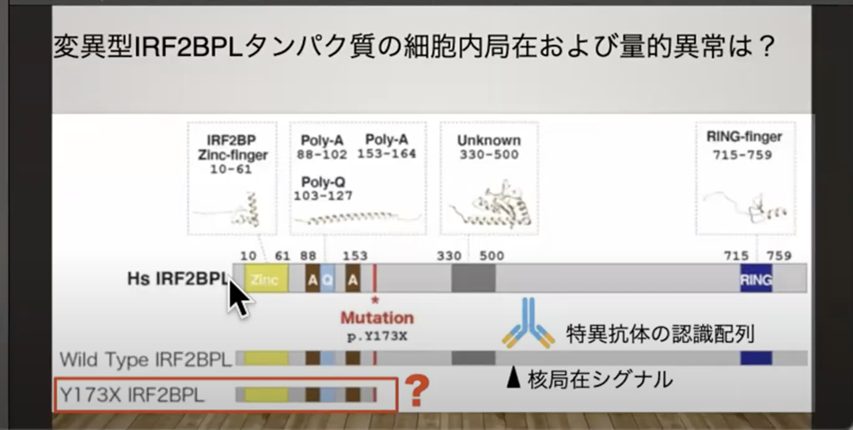
タンパク質は、遺伝子からアミノ酸の情報が転写され、アミノ酸が連なって形作られます。このアミノ酸の鎖が立体構造を形成することで、酵素としての機能を持つのです。
これが異常を起こすと、疾患が発症すると考えられています。
NEDAMSSの患者さんでは、IRF2BPL遺伝子に変異が入っていることがわかっています。通常、この遺伝子は父性と母性の2つのコピーを持ちますが、重症の症状であるNEDAMSSと呼ばれる疾患では、「ナンセンス変異」が発生します。
「ナンセンス変異」は、アミノ酸鎖に飜訳する途中で「止まれ」を意味するストップコドンが発生し、完全なタンパク質ができなくなる変異です。この異常が病気の原因となっています。
この短くなったタンパク質が、どのように症状の原因となるかは不明な点が残されています。しかし、理論的にはこの変異部分を正常な配列に書き換えれば、病気は治癒する可能性があります。実際、ストップコドンを無視してタンパク質の翻訳を継続させる薬剤はすでに開発されています。
また、遺伝子治療によりこの変異部分を直接修復する方法や、変異した配列のみを抑制する方法なども考えられています。遺伝子治療には様々な選択肢があり、今後さらに技術が進歩すると期待されています。
最終的には、小児科医が有望な治療法を見極め、患者さんに実際に投与するかどうかを判断する必要があります。
もう一つは、先ほどお話しした対処療法ではなく、実際に根本的な細胞レベルへのアプローチです。
具体的には、NEDAMSSが発症した際に見られる細胞の異常なシグナル伝達を正常化するような、薬理学的な処置を行います。
実はこの治療法は、既存の薬を使ってその病気を治療できないかという発想なので、実現可能性が高いと思っています。既に認可されていて安全性が確立されている薬を使うので、ハードルが低くなるんです。
「ドラッグリパーパシング」や「ドラッグリポジショニング」と呼ばれるこの手法は、今後の治療法の有望な選択肢の一つとして注目されています。
―――NEDAMSSの研究を進めていく中で、他の希少疾患研究に副次的にどんな影響を及ぼしていくと思いますか?
製薬企業は、一般的に患者数が少ない希少疾患や難病への新薬開発に積極的ではありません。いわゆる「アンメットニーズ」という状況で、莫大な開発費に見合う利益が見込めないためで、経営としては当然の判断だと言えます。そのため、原因はわかっても、実際に治療法が開発されないケースが多くありました。
しかし今回、ショウジョウバエの研究などを手がかりに、NEDAMSSという希少難病の原因遺伝子が特定されたことは、大きな突破口となる可能性があります。
特にアメリカでは、民間の財団が設立されるなど、NEDAMSSの治療法開発に向けた機運が高まっています。製薬企業による協賛は変わらず期待しにくい状況の中で、こうした民間レベルでの支援が活発化していくことは、とても良いことだと思います。
実際に日本でも、患者の家族から寄付を受けるなど、民間での助け合いの動きが芽生えつつあります。このような仕組みが整えば、希少疾患の基礎研究がさらに進展すると期待できます。
先日、四国地方のある家族から連絡がありました。長年、大学病院で娘さんの診断がつかず、原因不明のまま過ごしてきたそうです。
しかし、ようやく遺伝子検査でIRF2BPLに変異があることが判明したそうです。それ以上の情報は得られていないものの、少なくとも原因が特定できただけでも、家族にとっては大きな一歩だったはずです。
このように、診断がつかず苦しむ希少疾患の患者家族にとって、遺伝子変異が特定されただけでも、大きな前進だと思います。原因不明で途方に暮れていた状況から脱却できたことの意義は計り知れません。
絶対に諦めない、希少難病を解き明かす情熱

―――研究者としての使命感について、井上先生の見解をお聞かせください。
このNEDAMSSに関するプロジェクトは、私が研究を中断しても、病気自体は厳然と存在し続けますから、絶対に諦めるわけにはいきません。
確かに研究には、うまくいく時期とそうでない時期があります。通常の研究であれば、アイデアがうまくいかなければ、別のアプローチを試すため、前のアイデアは切り捨てていくものです。結果を出すためにはそうせざるを得ません。
従って今回の研究は成果を出すという観点からすれば、非常に非効率的なのが事実です。でもこの研究に関しては、「最後まで諦めてはいけない」と常に考えています。
きっかけは、最初にNEDAMSSを発症した少年と、その母親に出会ったことにあります。母親の強い願いを目の当たりにし、決してこの研究を放棄することはできなくなりました。
先ほども申し上げましたように、小児科医がこの病気を認知し、臨床課題として取り組んでくれるまでは、この研究を続けていこうという覚悟です。
―――最後に読者の皆様にメッセージをお願いします。
私一人の力だけでは、科学技術によってこの疾患を治せると断言できる立場にはありません。私は基礎研究に携わっていますが、実際にこの研究を前に進めるためには、小児科医の協力が不可欠です。このように、色々な人が集まって、力を合わせていくことが大切です。
そしてこの研究においては、患者さんの家族の強い願いが大きな原動力になっています。家族一人ひとりの「この病気を治したい」「世間に知らせたい」という思いを、少しずつ集め、人々を動かしていけたらと考えています。
患者さんやご家族の切実な想いに支えられながら、研究者、医師、それぞれが持つ力を合わせることで、着実に前進できると信じています。

ライター
So-gúd編集部
新井 那知
埼玉県・熊谷市出身。渋谷の某ITベンチャーに就職後、2016年にフリーランスライターとして独立。独立後は、アパレル、音楽媒体、求人媒体、専門誌での取材やコラム作成を担当する。海外で実績を積むために訪れたニューヨークで、なぜかカレー屋を開店することに—-帰国後は、クライアントワークを通してライターとして日々取材や編集、執筆を担当する。料理と犬、最近目覚めたカポエイラが好き(足技の特訓中)。
この記事を読んで、あなたは未来を変えたいと思いましたか?
はいいいえ
良かったらその未来をシェアしてくださいね!
はい
100%
いいえ
0%
良かったらその他の記事をご覧ください!
はい
100%
いいえ
0%
クリップボードにコピーしました。







